Genomics for forestry research

Genoinseq, Biocant's Advanced Sequencing Unit, has produced genomic data for forestry research. The Unit participated in the creation of several genomic resources, the most emblematic being the sequencing project of the cork oak transcriptome (Leal et al., 2014) and later the sequencing of the cork oak genome (Ramos et al., 2018), up to the sequencing of fungal and bacterial genomes (Leal et al., 2021, Margesin et al., 2018).
On the other hand, its activity in the area of transcriptomics has generated information to identify which genes are expressed in certain plant diseases, such as pine infection by the pine wood nematode (Santos et al., 2012, Figueiredo et al., 2013).
The recognition of the importance of microbial communities associated with plants, with the concept of the plant as an holobiont, led to the study of several epiphytic, endophytic and rhizospheric communities. These studies mainly targeting bacteria and fungi, identified microorganisms present in different communities, and studied their interaction with the plant and their role in the appearance of diseases (Pinho et al., 2020).
Discovery of molecular markers for the detection of geographic origin of the pinewood nematode and detection kit development
Bursaphelenchus xylophilus is the causal agent of pine wilt disease. The initial burst of the disease occurred in 1999, in Setúbal, Portugal, and, in spite of containment measures, the nematode spread to the entire country. The nematode was first identified in the United States, later in Asia and now in Portugal. The nematode infects pines when the insect vector, Monochamus, feeds in young twigs. Once inside the pine, the nematode reproduces and disperses in the xylem, causing cavitation, cessation of water flow and pine wilt.
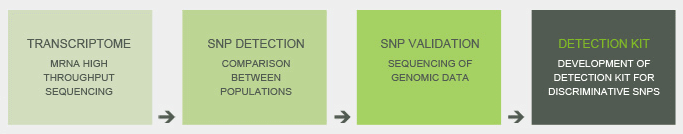
Through next generation DNA sequencing technologies, we sequenced the transcriptome of seven B.xylophilus isolates from four distinct geographic regions, four from Portugal, one from China, one from Japan and one from the United States. The diversity of transcripts sequenced was high; thus we just compared the sequences of genes involved in B.xylophilus pathogenicity, namely cellulases, pectin lyases, expansins and venom allergen proteins, and other pathways such as ubiquitination, and fungal feeding (chitinases), detecting 136 SNPs.
We validated the SNPs in corresponding genes in DNA in a wider set of isolates and used these polymorphisms to measure the genetic distance between isolates. This analysis revealed that Portuguese isolates are distant from United States and the Japanese isolates, are more closely related to Chinese isolates and identical to a Korean isolate. Among detected SNPs, five sufficed to discriminate between the four geographic locations. To ease the detection of the differentiating SNPs we developed a highly sensitive and efficient identification kit, based in real-time PCR.
Aquatic hyphomycete identification by ITS2 sequencing
Aquatic hyphomycetes play a pivotal role in organic matter turnover in headwater streams. These fungi produce extracellular enzymes that break down complex plant polymers and transform plant material into a more suitable and nutritious food source for invertebrate shredders.
The group of Fernandes and collaborators compared the efficiency of molecular methods namely DGGE (Denaturing Gradient Gel Electrophoresis) and 454 pyrosequencing of the transcribed spacer 2 region (ITS2) of the ribosomal RNA gene to the classical spore identification through microscopy to assess aquatic hyphomycete diversity on Quercus robur leaves decomposing in five Portuguese streams.
The researchers found that pyrosequencing was more efficient than DGGE to identify OTUs (Operational Taxonomic Units). However, only 53% of the hyphomycetes species were recovered from both spore morphology and pyrosequencing, while 26% and 21% were recovered only by spore morphology or only pyrosequencing, respectively.
Two problems were identified for hyphomycetes identification. One of the problems of pyrosequencing failure was the absence of ITS barcodes in the database for some of the species identified by spore morphology. Another problem was the 3% phylogenetic distance commonly used to generate OTUs. Some aquatic hyphomycete species that are easily distinguishable by spore morphology have ITS sequences differing by less than 3%. For instance, Lemonniera alabamensis and L. aquatica have ITS sequences differing by only 1.3% and even species belonging to different genera, such as Heliscus submersus and Flagellospora penicillioides, differ by only 1.1%.
Overall, 454 pyrosequencing is a powerful tool for revealing aquatic hyphomycete diversity, but there is a need to continually barcode fungal species ITS to cover all the extent of fungal diversity in the databases.
Fernandes I, Pereira A, Trabulo J, Pascoal C, Cássio F, Duarte S. 2015. Microscopy- or DNA-based analyses: Which methodology gives a truer picture of stream-dwelling decomposer fungal diversity? Fungal Ecology, 18:130-134.
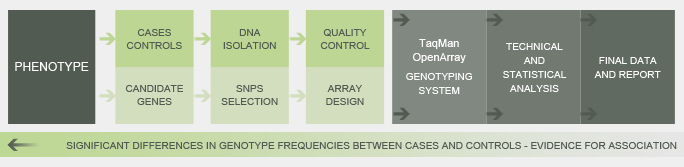
Case-Control Genetic Association Studies
Genetic association analysis has been performed in a case-control setting for identification of the genetic determinants of a certain phenotype. Significant differences in allele or genotype frequencies between cases (affected individuals) and controls (unaffected subjects) are taken as evidence for its involvement in the phenotypic trait under study.
These studies have been largely used for achieving insight into disease pathogenesis as a means of understanding the genetic basis of human diseases to improve preventive strategies, diagnostic tools and therapies. In addition, they have been increasingly applied for identifying genetic markers to select animals or plants based on marker assisted selection (MAS), whereby a trait of interest (e.g. productivity, disease resistance, abiotic stress tolerance) is selected based on genetic markers linked to it.
Using our TaqMan OpenArray Genotyping System, an association study was carried out by allelic discrimination of 741 SNPs within 51 candidate genes in 266 DNA samples from two groups of subjects with opposite phenotypic traits.
Concerning the DNA samples, 98.1% were successfully genotyped with an average call rate of 98%. In relation to the genotyping assays, 96.6% showed high genotyping quality being then analyzed for genotypic and allelic risk association tests. Significant differences in genotypic frequencies between cases and controls were observed for several SNPs, in two of the analyzed genes, providing strong evidence for their association with the phenotypic trait on study.