Genómica na investigação florestal

A Genoinseq, Unidade de sequenciação Avançada do Biocant, tem desde o início produzido dados genómicos para a investigação florestal. A Unidade participou na criação de vários recursos genómicos, os mais emblemáticos o projeto de sequenciação do transcriptoma do sobreiro (Leal et al., 2014) e mais tarde a sequenciação do genoma do sobreiro (Ramos et al., 2018), até à sequenciação de genomas de fungos e bactérias (Leal et al., 2021, Margesin et al., 2018).
Por outro lado, a sua atividade na área da transcritptómica tem gerado informação para identificar quais os genes expressos em determinadas doenças de plantas, por exemplo o processo de infeção do pinheiro pelo nematode-da-madeira-do-pinheiro (Santos et al., 2012, Figueiredo et al., 2013).
O reconhecimento da importância das comunidades microbianas associadas às plantas, com o conceito de planta como um holobionte, levou ao estudo de diversas comunidades epífitas, endófitas e da rizosfera, permitindo identificar os microrganismos presentes, por exemplo bactérias e fungos, estudar a sua interação com a planta e o seu papel no aparecimento de doenças (Pinho et al., 2020).
Descoberta de marcadores moleculares para determinação da origem geográfica do nemátode-da-madeira-do-pinheiro
Bursaphelenchus xylophilus é o nemátode causador da doença da murchidão do pinheiro. O foco inicial da doença ocorreu em Setúbal em 1999 e, apesar das medidas de contenção, espalhou-se ao país inteiro. O nemátode é originário dos Estados Unidos, pensa-se que foi difundido para a Ásia e daí para Portugal. O nemátode é introduzido nas árvores quando o inseto vetor, do género Monochamus, se alimenta dos ramos jovens. Uma vez dentro do pinheiro, reproduz-se no xilema, causando cavitação dos vasos, interrupção da passagem de água e murchidão.
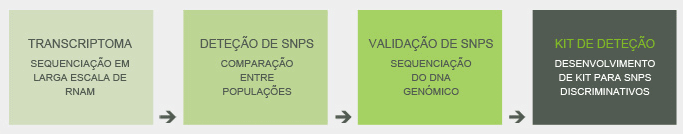
Recorrendo a tecnologias de sequenciação de última geração, sequenciámos o transcriptoma de sete isolados de quatro regiões distintas: quatro isolados portugueses, um isolado chinês, um isolado japonês e um isolado dos Estados Unidos. A comparação das sequências dos transcritos para genes de patogenicidade e de outras vias, nomeadamente celulases, liases de pectato, expansinas e proteínas alergénicas, proteínas envolvidas na ubiquitinação e quitinase, envolvida na alimentação a partir de fungos, revelou a presença de 136 SNPs.
A validação destes SNPs efetuou-se nos genes correspondentes, ao nível do DNA, num conjunto alargado de isolados. Os SNPs foram utilizados para aferir a distância genética entre os vários isolados, análise que revelou a distância dos isolados portugueses dos isolados americanos e japoneses, a proximidade com os isolados chineses e a similitude com um isolado coreano. Entre os SNPs encontrados foi possível selecionar um conjunto de cinco SNPs com capacidade para discriminar os quatro locais de origem. Para a deteção expedita destes SNPs diferenciadores foi desenvolvido um kit de identificação, baseado em PCR em tempo real, que permite determinar a origem geográfica do nemátode, de forma rápida e com elevada sensibilidade.
Identificação de hifomicetes aquáticos por sequenciação da região ITS2
Os hifomicetos aquáticos desempenham um papel essencial nos ciclos de matéria orgânica nas nascentes dos rios. Estes fungos produzem enzimas extracelulares que degradam os polímeros complexos de plantas numa fonte de material mais adequado e nutritivo para os invertebrados detritívoros.
O grupo de Fernandes e colaboradores comparou a eficiência da análise da região ITS2 através dos métodos moleculares DGGE (Eletroforese em gel com gradiente de desnaturação) e a pirosequenciação 454 com a identificação morfológica dos esporos por observação microscópica para o estudo da biodiversidade de hifomicetos em folhas de Quercus rubor em decomposição em cinco ribeiros portugueses.
Estes investigadores observaram que a pirosequenciação foi mais eficiente que DGGE na identificação de OTUs (Operational TAxonomic Units). Contudo, apenas 53% dos hifomocetos foram identificados em simultâneo por pirosequenciação e por morfologia de esporos, enquanto 26% foram identificados apenas por morfologia de esporos e 21% por pirosequenciação.
Dois problemas foram identificados para as diferenças obtidas. Um dos problemas está relacionado com a ausência de sequências de ITS nas bases de dados para algumas das espécies identificadas por morfologia. A outra questão prende-se com os limites de distância filogenética de 3% normalmente utilizados para a criação de OTUs na pirosequenciação. Alguns hifomicetos aquáticos com morfologia distinta nos esporos têm sequências de ITS que diferem menos de 3%. Por exemplo as sequências ITS de Lemonniera alabamensis e L. aquatica diferem apenas em 1,3%. Mesmo espécies pertencentes a géneros diferentes, como Heliscus submersus e Flagellospora penicillioides, diferem apenas em 1,1%.
De forma global a sequenciação 454 é uma ferramenta poderosa para o estudo da diversidade de hifomicetos aquáticos mas é necessário identificar um maior número de sequências de ITS para enriquecer a diversidade das bases de dados.
Fernandes I, Pereira A, Trabulo J, Pascoal C, Cássio F, Duarte S. 2015. Microscopy- or DNA-based analyses: Which methodology gives a truer picture of stream-dwelling decomposer fungal diversity? Fungal Ecology, 18:130-134.
Estudos de associação genética tipo caso-controlo
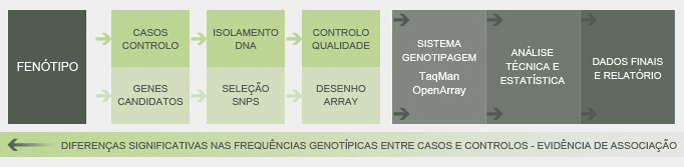
Estudos de associação genética têm sido realizados num contexto caso-controlo para identificação das determinantes genéticas de um certo fenótipo. Diferenças significativas nas frequências alélicas ou genotípicas entre casos (indivíduos afetados) e controlos (indivíduos não afetados) são tidas como evidências para a associação destes factores com a característica fenotípica sob estudo.
Estes estudos têm sido amplamente utilizados para melhor entender a patogénese genética de determinadas doenças afim de melhorar as estratégias preventivas, meios de diagnóstico e terapias. Este tipo de estudos têm sido também, cada vez mais, aplicados na identificação de marcadores genéticos para selecionar animais ou plantas (MAS, seleção assistida por marcadores), através dos quais uma característica de interesse (por exemplo, produtividade, resistência a doenças, tolerância a stress abiótico) pode ser selecionada.
Um estudo de associação foi realizado, com o nosso sistema de genotipagem TaqMan OpenArray, por discriminação alélica de 741 SNPs de 51 genes candidatos em 266 amostras de DNA correspondentes a dois grupos de indivíduos com características fenotípicas opostas.
No que respeita às amostras de DNA, 98.1% foram genotipadas com a qualidade requerida e um “call rate” médio de 98%. Em relação aos ensaios de genotipagem, 96.6% apresentaram a qualidade desejável sendo então considerados para a análise de associação de risco. Diferenças significativas nas frequências genotípicas nos casos em comparação com os controlos foram observadas para vários SNPs, em dois dos genes analisados, constituindo uma forte evidência para a sua associação com a característica fenotípica em estudo.